Vi förklarar MDR
Medicintekniska produkter används till nytta för patienter, för behandling eller diagnostik. Som sådana räknas allt från enkla plåster till komplexa medicintekniska produkter eller implantat som en konstgjord höftled. Tillverkare av medicintekniska produkter har varit tvungna att uppfylla lagliga och normativa krav under lång tid. I Tyskland regleras detaljerna i lagen om medicintekniska produkter. Den implementerar i sin tur europeiska krav som är nedskrivna i en europeisk förordning (Medical Device Regulation, förkortat MDR).
Varje land måste implementera de kraven i sina egna nationella lagar. I förordningen anges att en medicinteknisk produkt som släpps ut på marknaden inom EU måste uppfylla vissa krav. Vilka de är i detalj beror av vilka risker som kan uppkomma från den enskilda produkten. Ju mer komplex en produkt är, desto mer detaljerade är specifikationerna. För att göra det lättare att klassificera produkter tilldelas de till riskklasser.
För produkter med högre klassificering tillverkaren förse sitt anmälda organ (kontrollorgan) med detaljerade bevis på att alla krav uppfyllts. Därefter kan produkten få ett CE-märke och fritt marknadsföras var som helst i EU. För produkter med lägre risk (t.ex. klass I-produkter) får tillverkaren CE-märka på eget ansvar.
Den 26 maj 2021 ersattes det gamla europeiska direktivet MDD av en ny förordning, Medical Device Regulation, förkortat MDR. Till skillnad från direktiv får förordningar inte omarbetas till nationella lagar utan är omedelbart juridiskt giltiga och bindande för alla europeiska medlemsländer.
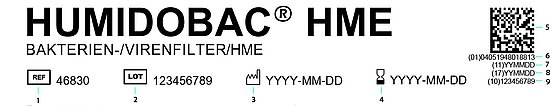
1: Artikelnummer
2: Batchnummer
3: Tillverkningsdatum
4: Utgångsdatum
5: QR-kod
6: GTIN (ID-nummer för produkt)
7: Tillverkningsdatum
8: Utgångsdatum
9: Batch
Du kommer inte ens att märka de flesta av förändringarna. Fokus ligger nämligen på interna processer och hur de organiseras. Nästan allt som rör MDR utspelas i bakgrunden. Men i och med att den nya förordningen träder i kraft i maj 2021 och att en tillhörande ny märkningsplikt, den så kallade UDI (Unique Device Identification) införs, kommer märkningen på våra produkter att ändras. Utöver den vanliga informationen som artikelnummer, artikelnamn eller tillverkare finns ett nytt 14-siffrigt nummer under en QR-kod.
Numret och koden innehåller all viktig information, så att varje medicinteknisk produkt kan spåras tillbaka i detalj. Den här entydiga, internationellt enhetliga identifiering av medicintekniska produkter över hela livscykeln ger tydliga fördelar: felfri och snabb registrering i produktionsprocessen, på lagret, under plockning och frakt. För dig som användare innebär det framför allt en fullständig spårbarhet av din produkt och därmed ytterligare transparens och säkerhet.
Vad betyder den nya MDR-förordningen för FAHL och hur genomförs åtgärderna organisatoriskt?
Vår kvalitetschef Christa Peters sammanfattar det i ett nötskal.
Företaget Andreas Fahl Medizintechnik-Vertrieb GmbH är glada över att möta de nya utmaningarna i MDR och välkomnar de nya riktlinjerna för högre patientsäkerhet. Självklart fortsätter vi att marknadsföra hela produktportföljen under de nya godkännandeprocedurerna. Vi har energiska team inom områdena kvalitetsledning och produktutveckling som vi fortlöpande utbildar så att de nya kraven kan implementeras på ett målinriktat sätt.
Vårt sedan länge anmälda organ är redan ackrediterat för revision enligt MDR och kommer att granska oss för första gången i enlighet med den nya förordningen under loppet av det här året. Som vanligt kommer patientsäkerheten att vara vårt fokus även i framtiden.


Vad är MDR?
MDR (Medical Device Regulation) är en ny EU-förordning för ökad patientsäkerhet. Det ersätter det gamla EU-direktivet MDD (Medical Device Directive). Förändringar som berör alla medicintekniska produkterbeskrivs på flera hundra sidor, men även sådana som formulerar nya specifikationer enbart för vissa produktgrupper. Sammanfattningsvis är dock alla tillverkare av medicintekniska produkter nu skyldiga att
- utöka dokumentationen av produktrelaterade data,
- intensifiera samarbetet med leverantörer och övervaka det mer,
- ändra tillordningen av produkter som t.ex. programvara till högre riskklasser,
- införa övervakning av produkterna på marknaden under hela produktens livscykel och som ett resultat regelbundet på nytt utvärdera produktens säkerhet.
När den förlängda övergångsperioden har löpt ut (för högre klassificerade produkter med ett giltigt MDD-certifikat) måste varje medicinteknisk produkt uppfylla kraven i MDR senast den 27 maj 2024. Vissa experter och kritiker ser en risk att vissa etablerade och beprövade produkter kan försvinna från marknaden på grund av de kraftigt ökade kraven med det nya rättsläget. Det gäller i synnerhet nischprodukter då tillverkarna är representerade på marknaden i hanterbar utsträckning, men som skulle föranleda samma ansträngning som en massprodukt om MDR infördes.