Wir stellen uns der MDR
Medizinprodukte werden eingesetzt, um Patienten zu therapeutischen oder diagnostischen Zwecken zu dienen. Als Medizinprodukt wird ein einfaches Pflaster aber auch ein komplexes medizinisches Gerät oder ein Implantat wie z. B. ein künstliches Hüftgelenk definiert. Für alle Medizinprodukte müssen deren Hersteller schon seit langem gesetzliche und normative Vorgaben erfüllen. In Deutschland sind die Details im Medizinproduktegesetz geregelt. Dieses wiederum setzt europäische Vorgaben um, die in einer europäischen Richtlinie (Medical Device Directive kurz MDD) niedergeschrieben sind.
Die Vorgaben aus dieser Richtlinie muss jedes Land für sich in eigenen nationalen Gesetzen umsetzen. In der europäischen Richtlinie wird beschrieben, dass ein Medizinprodukt, das in der EU in Verkehr gebracht wird, bestimmte Voraussetzungen erfüllen muss. Welche das im Einzelnen sind, hängt davon ab, welche Risiken von dem einzelnen Produkt ausgehen können. Je komplexer ein Produkt, desto detaillierter die Vorgaben. Um die Einordnung von Produkten zu erleichtern, werden diesen Risikoklassen zugeordnet.
Bei höher klassifizierten Produkten muss der Hersteller seiner Benannten Stelle (Prüfstelle) detailliert nachweisen, dass er alle Anforderungen erfüllt hat. Anschließend darf er sein Produkt mit einem CE-Zeichen versehen und es überall in der EU frei auf dem Markt anbieten oder anders gesagt „in den Verkehr bringen“. Für risikoärmere Produkte (z. B. Klasse I Produkte) liegt dies in der Eigenverantwortung des Herstellers.
Am 26. Mai 2021 wird die alte europäische Richtlinie MDD durch eine neue Verordnung, die Medical Device Regulation, kurz MDR ersetzt. Diese Verordnung muss anders als die Richtlinie nicht in nationalen Gesetzen umgesetzt werden, sondern ist sofort für alle europäischen Mitgliedsstaaten einheitlich rechtsgültig und bindend.
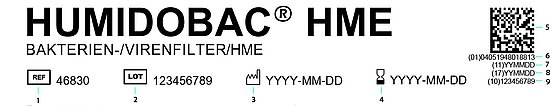
1: Artikelnummer
2: Chargennummer
3: Herstelldatum
4: Ablaufdatum
5: DataMatrix
6: GTIN
7: Herstelldatum
8: Ablaufdatum
9: Charge
Die meisten Veränderungen werden Sie gar nicht wahrnehmen. Der Fokus liegt nämlich auf internen Prozessen und deren Organisation. Fast alles bezüglich der MDR verläuft im Hintergrund. Mit Inkrafttreten der neuen Verordnung im Mai 2021 und der Einführung einer damit verbundenen neuen Kennzeichnungspflicht, der sogenannten UDI (Unique Device Identification), wird sich das Etikett unserer Produkte jedoch ändern. Zusätzlich zu den gewohnten Angaben wie beispielsweise Artikelnummer, Artikelname oder Hersteller findet sich dort eine neue 14-stellige Nummer unter einer scanbaren Datamatrix.
Die Nummer sowie die Matrix beinhalten alle wichtigen Informationen in einem scanbaren Code, um jedes Medizinprodukt im Detail zurückverfolgen zu können. Diese eindeutige, international einheitliche Identifikation medizinischer Produkte über den gesamten Lebenszyklus bietet deutliche Vorteile: fehlerfreies und schnelles Erfassen im Fertigungsprozess, im Lager, bei der Kommissionierung und im Versand. Für Sie als Anwender bedeutet es aber vor allem eine lückenlose Rückverfolgung Ihres Produktes und damit zusätzliche Transparenz und Sicherheit.
Was bedeuten die neuen MDR-Richtlinien für die Firma FAHL und wie werden diese Maßnahmen organisatorisch umgesetzt?
Unsere Qualitätsmanagerin Christa Peters bringt es kurz auf den Punkt.
Die Firma Andreas Fahl Medizintechnik-Vertrieb GmbH stellt sich diesen neuen Herausforderungen der MDR gerne und begrüßt die neuen Richtlinien für eine höhere Patientensicherheit. Natürlich bieten wir das gesamte Produktportfolio auch weiterhin unter den neuen Zulassungsverfahren am Markt an. Wir haben tatkräftige Teams im Bereich des Qualitätsmanagements sowie der Produktentwicklung, welche wir stetig weiterbilden, damit die neuen Anforderungen zielgerichtet umgesetzt werden können.
Unsere langjährige Benannte Stelle ist bereits für eine Auditierung nach MDR akkreditiert und wird uns im Laufe dieses Jahres erstmalig entsprechend der neuen Verordnung prüfen. Die Patientensicherheit steht für uns auch zukünftig wie gewohnt im Mittelpunkt.


Was ist die MDR?
Die MDR (Medical Device Regulation) ist eine neue Verordnung der europäischen Union für mehr Patientensicherheit. Sie ersetzt die alte Europäische Richtlinie MDD (Medical Device Directive). Auf mehreren hundert Seiten werden Veränderungen beschrieben, die alle Medizinprodukte betreffen, aber auch solche, die nur für bestimmte Produktgruppen neue Vorgaben formulieren. Zusammenfassend sind aber alle Hersteller von Medizinprodukten nun verpflichtet:
- die Dokumentation von produktbezogenen Daten zu erweitern,
- die Zusammenarbeit mit Zulieferern zu intensivieren und diese mehr zu überwachen,
- die Zuordnung von Produkten wie z. B. Software in höhere Risikoklassen zu verändern,
- die Überwachung der auf dem Markt befindlichen Produkte während des gesamten Lebenszyklus des Produktes einzuführen und resultierend daraus, die Sicherheit des Produktes regelmäßig neu zu bewerten.
Nach Ablauf der verlängerten Übergangsfrist (für höher klassifizierte Produkte mit gültigem MDD-Zertifikat) muss jedes Medizinprodukt spätestens zum 27.05.2024 den Vorgaben der MDR entsprechen. Darin sehen manche Fachleute und Kritiker die Gefahr, dass einige etablierte sowie bewährte Produkte aufgrund der deutlich erhöhten Anforderungen mit der neuen Gesetzeslage vom Markt verschwinden könnten. Das gilt insbesondere für Nischenprodukte, mit denen die Hersteller in überschaubarem Maß im Markt vertreten sind, die aber bei der Einführung der MDR den gleichen Aufwand verursachen würden wie ein Massenprodukt.