Wij gaan de uitdagingen van de MDR aan
Medische hulpmiddelen worden gebruikt om patiënten te behandelen of te diagnosticeren. Een medisch hulpmiddel wordt gedefinieerd als een eenvoudige pleister, maar ook als complex medische apparaat of een implantaat, zoals een kunstheup. Voor alle medische hulpmiddelen moeten de betreffende fabrikaten al sinds lange tijd aan wetten en normen voldoen. In Duitsland zijn de eisen gedetailleerd beschreven in het Medizinproduktegesetz. Dit is weer een omzetting van Europese eisen die in een Europese Richtlijn (de zogenaamde Medical Device Directive, kort MDD) worden beschreven.
De eisen in deze Richtlijn moet elk land in zijn eigen nationale wetten omzetten. In de Europese Richtlijn wordt aangegeven dat een medisch hulpmiddel dat in de EU in de handel wordt gebracht, aan bepaalde eisen moet voldoen. Welke eisen dat specifiek zijn, is afhankelijk van de risico's die het betreffende hulpmiddel kan veroorzaken. Hoe complexer een hulpmiddel, des te gedetailleerder de eisen. Om de classificatie van hulpmiddelen te vergemakkelijken, worden deze in risicoklassen onderverdeeld.
Bij hoger geclassificeerde hulpmiddelen moet de fabrikant zijn aangemelde instantie (keuringsinstantie) gedetailleerd bewijzen dat aan alle eisen wordt voldaan. Aansluitend mag de fabrikant de CE-markering op het hulpmiddel aanbrengen en het overal binnen de EU vrij op de markt aanbieden of anders gezegd 'in de handel brengen'. Bij hulpmiddelen die weinig risico vormen (bijv. hulpmiddelen in klasse I) is de fabrikant zelf daarvoor verantwoordelijk.
Op 26 mei 2021 werd de oude Europese Richtlijn MDD vervangen door een nieuwe verordening, de zogenaamde Medical Device Regulation, kort MDR. In tegenstelling tot de richtlijn hoeft deze verordening niet in nationale wetten te worden omgezet, maar is direct voor alle Europese lidstaten uniform rechtsgeldig en bindend.
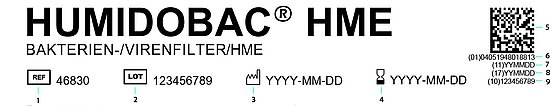
1: Artikelnummer
2: Partijnummer
3: Productiedatum
4: Datum waarop het certificaat zijn geldigheid verliest
5: Datamatrix
6: GTIN
7: Productiedatum
8: Datum waarop het certificaat zijn geldigheid verliest
9: Partij
De meeste veranderingen zult u helemaal niet kunnen zien. De nadruk ligt namelijk op interne processen en de organisatie daarvan. Bijna alles met betrekking tot de verordening medische hulpmiddelen (MDR) vindt op de achtergrond plaats. Met het van kracht worden van de nieuwe verordening in mei 2021 en de invoering van een daarmee verbonden nieuwe markeringsplicht, de zogenaamde UDI (Unique Device Identification - unieke hulpmiddelidentificatie), verandert echter het etiket op onze producten. Naast de gebruikelijke gegevens zoals artikelnummer, artikelnaam of fabrikant wordt een nieuw 14-cijferig nummer vermeld onder een scanbare datamatrix.
Het nummer en de matrix bevatten alle belangrijke informatie in één scanbare code, zodat elk medisch hulpmiddel gedetailleerd kan worden getraceerd. Deze eenduidige, international uniforme identificatie van medische hulpmiddelen gedurende de gehele levenscyclus biedt duidelijke voordelen: foutvrije en snelle registratie in het productieproces, in het magazijn, bij orderpicking en bij de verzending. Voor u als gebruiker betekent het echter vooral een naadloze tracering van uw product en dus extra transparantie en zekerheid.
Wat betekent de nieuwe MDR voor de firma FAHL en hoe zullen we deze maatregelen organisatorisch in de praktijk brengen?
Onze kwaliteitsmanager Christa Peters legt dit kort uit.
De firma Andreas Fahl Medizintechnik-Vertrieb GmbH gaat deze nieuwe uitdagingen van de MDR graag aan en verwelkomt de nieuwe richtlijnen voor meer veiligheid voor patiënten. Natuurlijk blijven we het gehele productassortiment ook volgens de nieuwe goedkeuringsprocedures aanbieden. Wij hebben geweldige teams voor kwaliteitsmanagement en productontwikkeling, die wij continu bijscholen zodat we doelgericht aan de nieuwe eisen kunnen voldoen.
Onze jarenlange aangemelde instantie is reeds geaccrediteerd voor audits volgens de MDR en zal onze producten in de loop van dit jaar voor het eerst volgens de nieuwe verordening controleren. De veiligheid van patiënten blijft voor ons ook in de toekomst zoals gebruikelijk centraal staan.


Wat is de MDR?
De MDR (Medical Device Regulation - Verordening betreffende medische hulpmiddelen) is een nieuwe verordening van de Europese Unie gericht op meer veiligheid voor patiënten. Deze verordening vervangt de oude Europese Richtlijn MDD (Medical Device Directive - Richtlijn betreffende medische hulpmiddelen). Op meerdere honderd pagina's worden veranderingen beschreven die gelden voor alle medische hulpmiddelen, maar ook veranderingen die alleen aan bepaalde productgroepen nieuwe eisen stellen. Samenvattend zijn echter alle fabrikanten van medische hulpmiddelen verplicht om:
- de documentatie van productgerelateerde gegevens uit te breiden;
- de samenwerking met toeleveranciers te intensiveren en deze meer te bewaken;
- de toewijzing van producten zoals software aan hoge risicoklassen te veranderen;
- de in de handel gebrachte producten tijdens de gehele levenscyclus van het hulpmiddel te bewaken en als gevolg daarvan de veiligheid van het hulpmiddel regelmatig opnieuw te beoordelen.
Na afloop van de verlengde overgangstermijn (voor hoger geclassificeerde hulpmiddelen met geldig MDD-certificaat) moet elk medisch hulpmiddel uiterlijk op 27.05.2024 aan de eisen van de MDR voldoen. Sommige experts en critici zien daarin het gevaar dat enkele gevestigde en bewezen producten vanwege de duidelijk hogere eisen vanwege de nieuwe wetgeving van de markt zouden kunnen verdwijnen. Dat geldt met name voor nicheproducten, die fabrikanten op overzichtelijke wijze op de markt brengen, die echter bij de invoering van de MDR dezelfde kosten en inspanningen zouden veroorzaken als een massaproduct.


